Overview¶
CoVigator dashboard: https://covigator.tron-mainz.de
Human infections with SARS-CoV-2 are spreading globally since the beginning of 2020, necessitating preventive or therapeutic strategies and first steps towards an end to this pandemic were done with the approval of the first mRNA vaccines against SARS-CoV-2. The accumulation of virus samples that have been sequenced in a short time frame is unprecedented (see Figure 1). This is the first pandemic recorded at a molecular level with such level of detail giving us the opportunity to develop new tools for the monitoring of its evolution.
We want to provide an up-to-date interactive view on SARS-CoV-2 mutations to support global efforts in preventing or treating infections. Monitoring the appearance of relevant new mutations is key to enable a fast reaction to new strains and for that purpose we enable the exploration of these mutations and their annotations (see Figure 2). Thus, we envision to help guiding global vaccine design efforts to overcome the threats of this pandemic.
CoVigator is a monitoring system for SARS-CoV-2 which integrates a full variant calling pipeline, a database that stores all relevant information about mutations in SARS-CoV-2 and finally a dashboard to enable visual analytics.
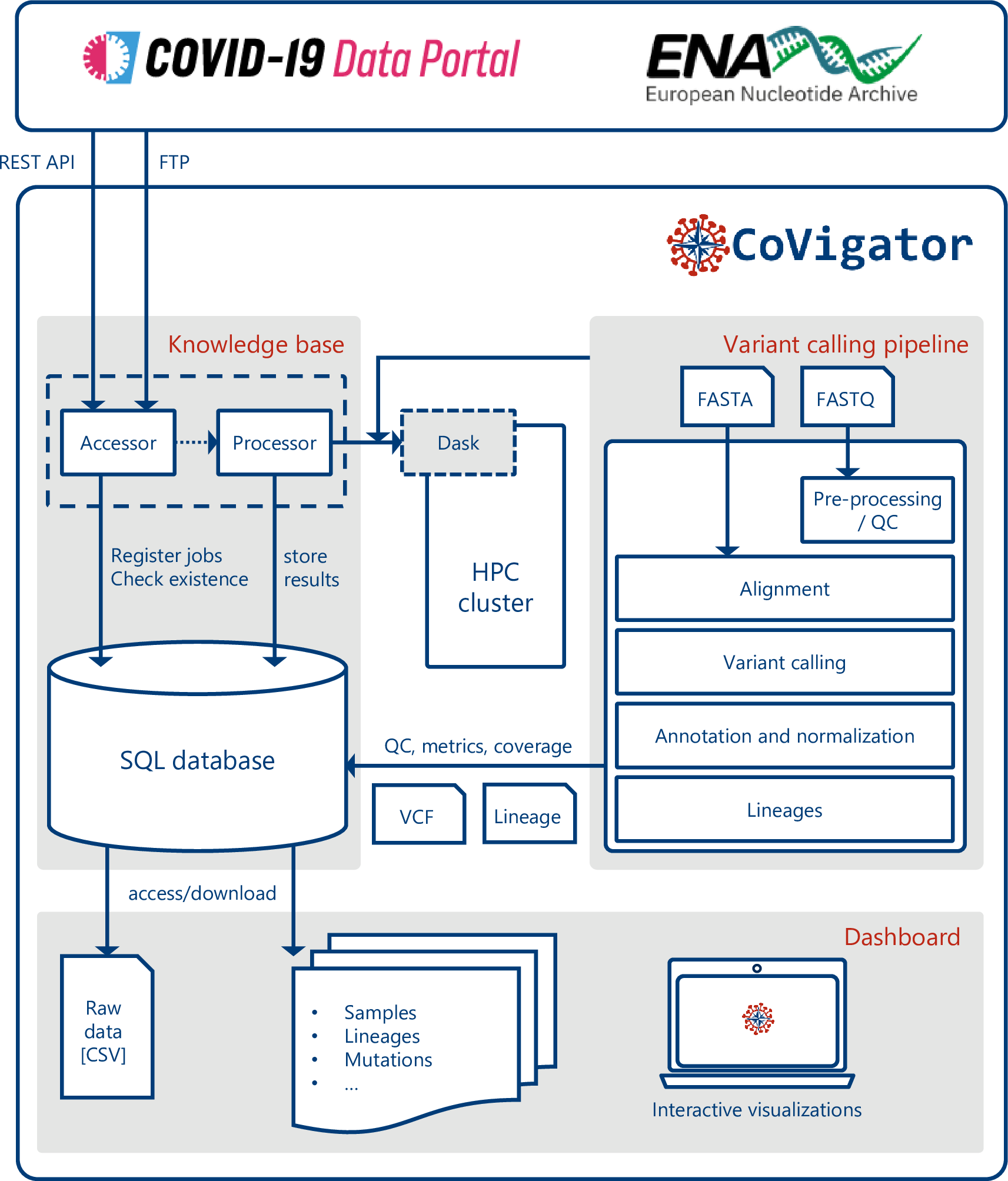
Figure 1: CoVigator system components. The accessor reads external data and stores it in an SQL database. The processor reads the stored data and distributes the processing of every sample in an HPC cluster via Dask. The pipeline processes FASTA and FASTQ data and finally stores the results back in the database. The dashboard reads the results and displays them in a set of interactive plots. The results are also available in raw format.

Figure 2: Samples by country tab plots for raw reads dataset. A) accumulation of samples through time by country; B) dN/dS ratio through time on each SARS-CoV-2 protein; C) dN/dS ratio through time in the domains of the spike protein.
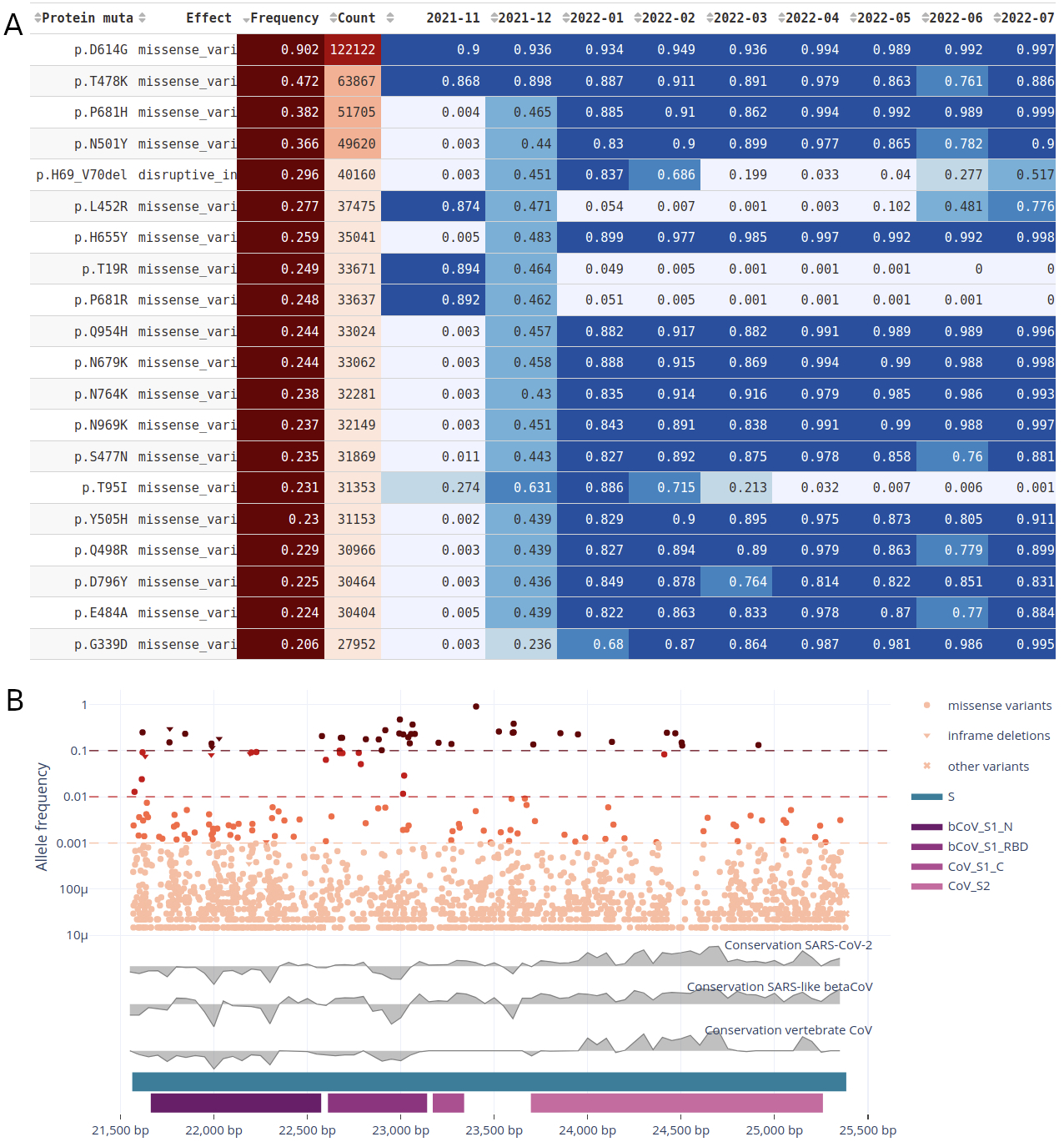
Figure 3: Gene view for the spike protein on the raw reads dataset. A) Table of the top 20 recurrent mutations with the frequency segregated by month between November 2021 and July 2022; B) gene view showing mutations (synonymous and unique mutations excluded) in the spike protein and their frequencies in the virus population, the ConsHMM conservation tracks in grey and the Pfam protein domains in tones of purple.
CoVigator loads publicly available SARS-CoV-2 DNA sequences from two databases:
European Nucleotide Archive (ENA) providing raw reads in FASTQ format.
COVID-19 Data Portal providing assemblies in FASTA format.
There is certain overlap in the samples present in ENA and COVID-19 Data Portal as some national initiatives are systematically reporting both FASTQ reads and FASTA assemblies. FASTQ reads enable a higher resolution into the SARS-CoV-2 mutation details through the individual reads. This allows us to annotate mutations with a Variant Allele Frequency (VAF) and explore intrahost mutations. On the other hand, while we load all of the sequences from COVID-19 Data Portal database in CoVigator, we only process the Illumina samples from ENA. This means excluding all of the Oxford Nanopore samples and hence having a partial view of all the available data. Each of the datasets is available in a separate address https://covigator.tron-mainz.de/covid19-portal and https://covigator.tron-mainz.de/ena, respectively.
The dashboard is implemented in the visualization framework Dash. The computation is distributed through our cluster with a library of similar name Dask. The analysis pipeline is implemented in the Nextflow framework.
The CoVigator project was developed at the Biomarker Development Center at TRON (Translational Oncology at the University Medical Center of the Johannes Gutenberg University gGmbH). The project was kindly supported by Intel´s Pandemic Response Technology Initiative.
How to cite¶
Bukur, T., Riesgo-Ferreiro, P., Sorn, P., Gudimella, R., Hausmann, J., Rösler, T., Löwer, M., Schrörs, B., & Sahin, U. CoVigator — A Knowledge Base for Navigating SARS-CoV-2 Genomic Variants. Viruses. 2023; 15(6):1391. 10.3390/v15061391
Schrörs, B., Riesgo-Ferreiro, P., Sorn, P., Gudimella, R., Bukur, T., Rösler, T., Löwer, M., & Sahin, U. (2021). Large-scale analysis of SARS-CoV-2 spike-glycoprotein mutants demonstrates the need for continuous screening of virus isolates. PLOS ONE, 16(9), e0249254. 10.1371/journal.pone.0249254
Open source¶
All the code for CoVigator is open source and made available under the MIT license. We welcome any contribution in any of our code repositories. If you have trouble using CoVigator or you find an issue, we will be thankful if you would report a ticket in GitHub.
Our repositories:
CoVigator knowledge base and dashboard: https://github.com/TRON-Bioinformatics/covigator
CoVigator analysis pipeline: https://github.com/TRON-Bioinformatics/covigator-ngs-pipeline
CoVigator data analysis playing ground: https://github.com/TRON-Bioinformatics/covigator-analysis
About TRON¶
TRON is an independent biopharmaceutical non-profit translational research organization pursuing new diagnostics and drugs for the treatment of cancer and other diseases with high medical need. We focus our transdisciplinary competencies in genomics and immunology to 1) develop novel platforms for the identification and validation of “omics”-based biomarkers and 2) for harnessing and 2) modulating immune system components, for use in personalized therapies. Partnering with academia and industry, TRON executes research at the leading edge to support innovative drug design for human health.
Acknowledgements¶
Intel is committed to accelerating access to technology that can combat the current pandemic and enable scientific discovery that better prepares our world for future crises. Funding for this solution was funded in part by Intel’s Pandemic Response Technology Initiative. For more information about healthcare solutions from Intel, visit intel.com/healthcare. For more information about Intel’s COVID-19 response, visit intel.com/COVID-19.
We thank Franziska Lang and Özlem Muslu for critical discussions and feedback. We thank Rudolf Koopmann for his contribution to integrate Pangolin into the CoVigator pipeline.
We gratefully acknowledge all data contributors, i.e. the Authors and their Originating laboratories responsible for obtaining the specimens, and their Submitting laboratories for generating the genetic sequence and metadata and sharing via the European Nucleotide Archive [1] and the COVID-19 Data Portal [2], on which this research is based.
Leinonen, R., Akhtar, R., Birney, E., Bower, L., Cerdeno-Tárraga, A., Cheng, Y., Cleland, I., Faruque, N., Goodgame, N., Gibson, R., Hoad, G., Jang, M., Pakseresht, N., Plaister, S., Radhakrishnan, R., Reddy, K., Sobhany, S., Hoopen, P. Ten, Vaughan, R., Zalunin V., Cochrane, G. (2011). The European nucleotide archive. Nucleic Acids Research, 39(SUPPL. 1), D28. 10.1093/nar/gkq967
“COVID-19 Data Portal - Accelerating Scientific Research through Data.” Accessed October 24, 2022. https://www.covid19dataportal.org/.
The lineage annotation in the covigator dashboard package uses “the description of constellations of mutations for the SARS-CoV-2 virus” by cov-lineages provided and licensed under CC BY-NC 4.0
A note on terminology¶
There is some confusion around the terms variant and mutation. Variant may refer to a virus strain, e.g.: the alpha variant; but it may also refer to a genetic variant. A virus strain may have multiple genetic variants. We have decided to use the term mutation instead of variant to refer to a genetic variant. But exceptionally, we have kept the use of variant in some scientific terms commonly used; these are:
Variant Allele Frequency (VAF): the ratio of reads supporting a given mutation
Variant calling: the process by which all reads overlapping a given position are evaluated to determine whether a mutation may exist.
Variant call: a mutation determined by the variant calling process
Single Nucleotide Variant (SNV): a point mutation where a single DNA base is substituted by another
Multi Nucleotide Variant (MNV): a point mutation where more than one DNA base is substituted by another
There are two terms referring to a given mutation frequency:
The Variant Allele Frequency (VAF) refers to the ratio of reads supporting a given mutation. The VAF can only be calculated on the ENA dataset. The VAF is used to distinguish clonal and intrahost mutations.
The mutation frequency on the other hand refers to the frequency in the population of samples. This is calculated on both datasets ENA and COVID-19 Data Portal, but importantly intrahost mutations are not taken into account.